Data Integrity in Pharma: Why it Matters & How to Minimize it?
Maintaining data integrity is essential for the smooth running of an organization, particularly in the Pharma biotech sector. Ensuring the accuracy and consistency of datasets as they are produced, processed and published, not only guarantees an efficient internal process but also keeps companies on the right side of the law when it comes to data integrity in Pharma.
Since 2010, when the US Food and Drug Administration (FDA) incorporated data integrity into its primary inspection objectives, the number of data integrity warning letters issued in the Pharma biotech sector has increased substantially. A higher level of scrutiny will lead to more errors being detected, but nearly a decade later many companies still struggle to comply with these regulations.
Four causes of data integrity issues in Pharma:
1. Ignorance and disengagement
Pharma biotech employees can often fail to understand the importance of the system set out to maintain data integrity. Therefore they feel disengaged from the process and fall into a pattern of mindless box-ticking and failing to thoroughly assess whether or not data integrity has been maintained.
As a result, it’s vital for staff training sessions on data integrity to highlight the consequences. If the FDA finds out data integrity has not been maintained, it can lead to wide range of penalty from a formal warning letter to criminal prosecution and ought not to be taken lightly.
2. Company culture
Data integrity lapses can also occur as a result of poor corporate morale. A company culture is where employees feel unable to come forward and admit their mistakes can often lead to an increase in errors. If employees feel pressured to generate perfect data documents this can ironically, lead to data integrity issues. As disregard factors such as authenticity, accuracy and timeliness in favor of surface-level record keeping.
This can be mitigated through a working environment of openness and transparency. Logistical errors which could lead to data integrity breaches ought to be addressed as opportunities for learning and improvement, rather than punishment.
3. Human error
The human element, of course, can be hard to overcome. Even the most diligent employees can offer only the best they can, with the resources. Stress, fatigue and distraction can all lead to errors and thus data integrity issues.
The best way to work around this is to once more encourage a positive working environment, where employees feel valued both as members of an organization and as human beings. Happy workers will consistently deliver higher standards, not only in terms of data integrity but in all areas.
4. Inefficient systems
Finally, it’s important to note that complex data integrity systems will magnify the risk of problems in this area. Standard operating procedures should be simple and easy to navigate so that they remain fit for purpose, yet avoid falling into the trap of implementing any well-intentioned shortcuts which could allow data integrity breaches to slip through the net.
This is a delicate balance for any Pharma biotech company to strike and will be quite unique to each organization. As such, it is important for senior staff members to take their time to ensure the system of data integrity are rigorous and efficient.
How Data Integrity can be improved?
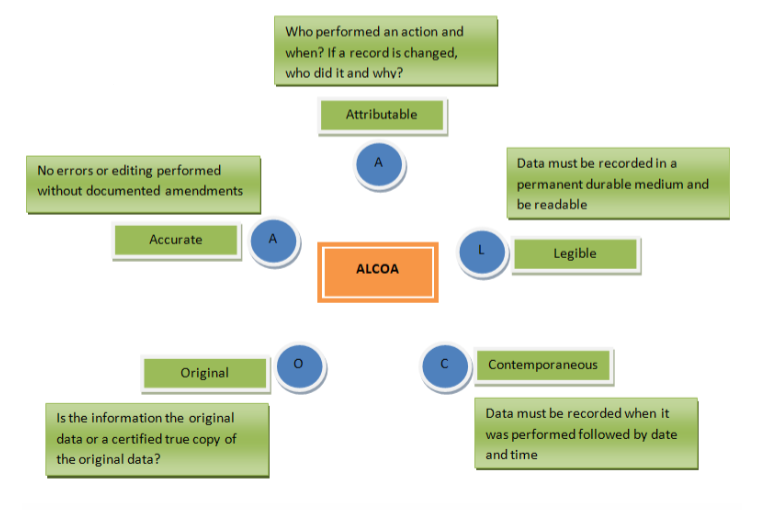
Data integrity & what it means to have ALCOA data
Attributable:
The main controls needed to maintain an attributable electronic record for the use of secure and unique user logins
and electronic signatures. Using generic login-IDs or sharing credentials should always be avoided. Unique user
logins allows individual to be linked to the creation, modification or deletion of data within the record. For a
signature to be legally-binding, there should be a secure link between the person signing and the resulting
signature. The use of digital images of handwritten signatures is not often considered a secure method for
electronically signing documents. These images lose their credibility when not stored in a secure location or when
they appear on documents that can be easily copied by others.
Legible:
In order for an electronic record to be considered legible, traceable and permanent it must utilize controls such as
writing SOPs and designing a system that promotes saving electronic data in concurrence with the execution of
the activity. This is best done by prohibiting the creation or manipulation of data in temporary memory as well as
immediately committing data to a permanent memory before moving on. Secure time-stamped audit trails should
be used to record operator actions. The system configuration should limit the enhanced security rights of users
such as turning off the audit trail or overwriting data. These administrative rights should be reserved (whenever
possible) for individuals who are independent of those responsible for the content of the electronic records.
Improperly overwriting data or manipulating the audit trail impairs the legibility of the data by obscuring the
original value of the record. This is equivalent to the use of single-line cross-outs in paper records to denote
changes to the data. The data in these paper records are changed but the original values must still be legible.
Contemporaneous:
Contemporaneous recording of data also utilizes the controls of writing SOPs and maintaining settings that
immediately commit data to permanent memory. In order for the data to be considered contemporaneous, the
record must also have a secure time stamp system that cannot be altered by the user. Time and date stamps should
be synchronized across all systems involved in the GxP activity. These controls should be true for both the
workstation operating system and any relevant software application used. Data is not considered contemporaneous
when recorded on an unofficial document and then later entered into the official electronic record.
Original:
Original electronic records (or certified true copies) should undergo review and approval procedures. These
reviews should describe the review method itself as well as any changes made to the information in the original
records. These include changes documented in audit trails or any other relevant metadata. Written procedures
should define the frequency, roles and responsibilities and approach to the review of metadata. A risk-based
approach to the scope of these procedures is recommended. Once reviewed, electronic data sets should be
electronically signed to document their approval.
Controls should also be put in place to guarantee the retention of original electronic documents as best as possible.
The original record should be routinely backed up and stored separately in a safe location. Secure storage areas
should have a designated electronic archivist who is independent of the GxP operation. Tests should be carried out
at times in order to verify that the copy can be retrieved and utilized from secure storage areas.
Accurate:
Data accuracy should be maintained through a quality management system that is risk-based and appropriate to
the scope of the operation. Routine calibration and equipment maintenance should be performed. Computer
systems that generate, maintain, distribute or archive electronic records should be validated. The entry of critical
data such as high priority formulas for spreadsheets should be verified by two authorized individuals. Once
verified, critical data fields should be locked to prevent modification by any unauthorized individuals.

Complete:
All information related to recording the data, including all changes / corrections to data, additional readings,
metadata and errors associated with the data collection should be retained. For critical manufacturing / processing
steps, all information to re-create the actions should be recorded.
Data and Metadata to be recorded should be defined in procedures or specifications and checks performed to
ensure no omissions. Manual checks, automated verification methods and validation can be used to ensure all the
required data and metadata is properly recorded. Procedures should clearly define critical processing steps and
records created to enable the steps and associated events to be created.
Consistent:
During its lifecycle, data must be protected from changes that may alter or corrupt the content and meaning of the
data or associated metadata. Data may be migrated to different media or systems (e.g. paper to electronic media,
and database migrations) provided the reason is documented and justified. The content, meaning and dynamic
nature of data should remain consistent throughout the lifecycle of the data.
In cases of multiple instances of electronic data, these should be synchronized and the primary instance defined.
For critical processing steps, the date and time should be recorded with the data to chronologically relate the
events/ information and corroborate the sequence.
Enduring:
Processes should be in place to ensure the data is preserved for the entire retention period. Paper and electronic
data should be stored and maintained in a manner to protect from degradation or loss and electronic data protected
from hardware/software obsolescence.
Available:
Paper and electronic data should be available for onsite review by regulatory authorities and authorized staff
throughout the required retention period. For remotely stored electronic data, a method for accessing and viewing
the data onsite should be available. Systems should be capable of generating validated electronic copies of
electronic data/ records for regulatory inspectors.